History
This is an eighteen-month old child born of Mexican extraction resident in Texas, USA . His prenatal and natal histories are unremarkable.
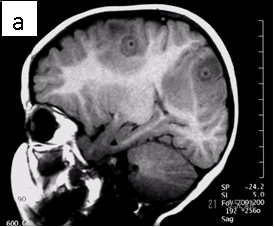
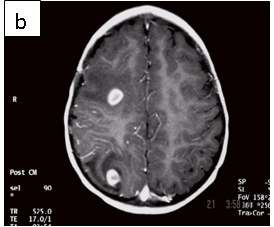
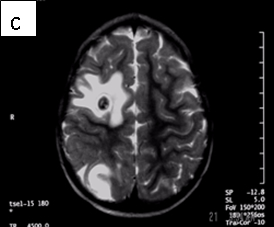
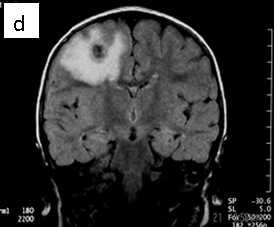
Figure 1: Neurocysticercosis in an 18-month child. a. Sagittal T1 without gadolinium; b. Axial T1 with gadolinium; c. Axial T2; d. Coronal FLAIR
Radiologic diagnosis: Neurocysticercosis.
Radiologic differential diagnosis
1. Pyogenic abscesses.
2. Tuberculomas.
3. Metastases.
4. Tuberous sclerosis.
5. Toxoplasmosis.
Discussion
Neurocysticercosis results when encysted larval forms of Taenia solium (pork tapeworm) invade the central nervous system (2). It is the most common parasitic disorder of the CNS (5) occurring in 4% of autopsy series from endemic countries of the world (in Latin America, Asia, Africa and some European countries).
| SPECIES | STAGE SEEN IN MAN | COMMON NAME | DISEASE IN MAN |
| Taenia saginita | Adult | Beef tape worm | Rarely symptomatic |
| Taenia solium | AdultLarva | Pork tape wormCysticercosis | Rare symptomaticCysts in the brain and other tissues |
| Taenia multiceps | Larva | Bladder worm, coenurosis | Cysts in the brain and eyes |
| Echinococcus granulosus | Larva | Hydatid cyst disease | Solitary tissue cysts |
| Echinococcus multilocularis | Larva | Alveolar cyst disease | Multilocular cysts |
| Diphyllobotrium latum | Adult | Fish tape worm | Pernicious anemia |
| Hymenolepis nana | Adult | Dwarf tape worm | Rarely symptomatic |
| Spirometra mansonoides | Larva | Sparganosis | Subcutaneous larvae |
Taenia solium is one of the eight cestode species that infect man (see Table 1). Humans are the definitive host for the parasite (we harbor the adult worm) while omnivorous or herbivorous vertebrates (like pig) host its larval form (intermediate host). Although the adult forms of the cestodes that we host rarely cause us harm, humans suffer variable degrees of illnesses when they host the larval forms; in the former case the parasites often reside harmlessly in the bowels, while in the latter they invade the tissues. For example, in its normal life cycle, humans harbor the pork tapeworm and shed its eggs (either individually or in proglottides impregnated with eggs) in our feces. The pig, an omnivore and its natural intermediate host, consumes the eggs of the parasite, which hatch into larval forms in its alimentary tract and invade its tissues. Consumption of undercooked meat from infected pork by us completes the cycle, freeing the encysted larvae when they encounter our gastric acid and bile salts. In contrast, we act as the intermediate host of the parasite when we ingest the tapeworm eggs by eating food infected with them (the infant’s mother in this case may have infected her child this way). Their larvae invade the bowel walls and invade our tissues as far afield as the brain, eyes, muscles, heart and other places wreaking havoc in their wake (7). In the CNS, the cysticerci (the larvae) may lodge in the brain parenchyma, the spinal cord, the subarachnoid space or the ventricles, lying dormant for years or causing various categories of clinical disease (see Table 2) (3).
| SYNDROME | CLINICAL FEATURES |
| Acute invasive stage | Commoner in children; mortality, 10%. Occurs immediately after the infection with fevers, headaches, and myalgias. Heavy infection may result in coma with rapid deterioration (cysticercal encephalitis). Treat aggressively with anti-parasitic and anti-inflammatory agents. |
| Parenchymal CNS cysticercosis | 50% of cases; there is established parenchymal disease with seizures, focal deficits, intellectual impairment, personality changes and signs of raised ICP when severe. |
| Subarachnoid cysticercosis | 30% of cases; there is larval invasion of the subarachnoid space including the cisterns causing disturbed CSF flow, sensorial changes and, as in other chronic basal meningitis, signs of vasculitis and parenchymal infarctions. |
| Intraventricular cysticercosis | 15% of cases; there are cysts in the ventricles, the 4th most frequently involved, causing intermittent obstruction to CSF flow with head movements. The aggressive form is racemose cysticercosis in which sheets of parasites spill out into the subarachnoid space severely impairing CSF drainage. It is commoner in young women |
| Spinal cysticercosis | Cord compression with radiculopathy, transverse myelitis and signs of meningitis. |
| Ocular cysticercosis | Eye pain with scotomata, iridocyclitis, clouding of the vitreous and retinal inflammation or detachment |
However, 2 to 10 years after CNS invasion, the dormant cysts may die, lose osmoregulation, absorb fluid and disintegrate, releasing antigens that set up variable degrees of inflammation. The clinical conundrum that results from CNS larval invasion depends upon the size of the invasion, and the location and degree of the inflammation. You can make a sure diagnosis of neurocysticercosis by analyzing infected tissue microscopically. But you can make a presumptive diagnosis of the disease if your patient is from or resides in an endemic area (as in this case) and, if laboratory analysis of their CSF specimen including an immunoblot test, and their CNS imaging results are positive for markers of the disease (4,5) (see Table 3).
| CSF analysis | Hypoglycorrhachia. Elevated CSF protein. Lymphocytosis and eosinophylic pleocytosis (5-500 cells/microliter). ELISA and Western blot testing for specific IgM and IgG anticysticercal antibodies in the CSF (75 to 100% sensitivity). |
| CT scan | A variable appearance including multiple, low-density lesions, 0.5 to 2.0cm in diameter. In acute disease, they enhance after contrast administration, surrounded by vasogenic edema with or without mass effects. Dead parasites (not dying parasites) show as non-enhancing calcified 5mm bodies. |
| MRI | Live forms have a characteristic appearance: fluid-filled lesions containing an inverted scoleces, surrounded by thin low-signal capsule. They do not stimulate inflammation and do not enhance; dying forms do. In the less common racemose type, the cysts may be hard to see because they have similar imaging features as the CSF. |
For cysts that cause symptoms outside the CNS, surgical resection achieves cure. The treatment of symptomatic neurocysticercosis, which carries a 50% mortality rate, is more problematic. Two drugs, albendazole and praziquantel control symptoms and cause regression in the size and number of cysts in patients with viable (non-enhancing) cysts in their brain parenchyma. However, they provide limited improvement in patients with arachnoiditis and none in patients with intraventricular cysts. These latter patients should be treated with surgery or palliated with ventricular shunting, anticonvulsants, and anti-inflammatory drugs. According to our case’s physician, he did not receive anti-parasitic medication because the imaging features suggested that the parasites were dying (vasogenic edema and ring enhancement) (6).
Two cautionary injunctions about treatment, though: first, 20% of patients with parenchymal cysticercosis worsen symptomatically following the institution of drug treatment as the parasites die and release their antigens. Concomitant administration of anti-inflammatory drugs subdues this phenomenon. Second, because anti-inflammatory agents alter the CNS pharmakokinetics of the anti-parasitic agents, their routine use is discouraged.
You should rescan your patients 3 months after therapy to judge their response to treatment; prescribe an alternate drug to the one used ab initio if there is no response.
For patients with ocular cysticercosis (remember, 20%), you are better off holding drug treatment until resection of their lesions because they do not respond well to medication.
Bibliography
1. Shandera WX, White AC Jr, Chen JC, Diaz P, Armstrong R. Neurocysticercosis in Houston, Texas. A report of 112 cases. Medicine (Baltimore). 1994 Jan;73(1):37-52.
2. Domenici R, Matteucci L, Meossi C, Stefani G, Frugoli G. Neurocysticercosis: a rare cause of convulsive crises Pediatr Med Chir. 1995 Nov-Dec;17(6):577-81. Italian.
3. Caparros-Lefebvre D, Lannuzel A, Alexis C, Strobel M, Janky E Cerebral cysticercosis: why it should be treated. Presse Med. 1997 Nov 1;26(33):1574-7. French.
4. Ruiz-Garcia M, Gonzalez-Astiazaran A, Rueda-Franco F. Neurocysticercosis in children. Clinical experience in 122 patients. Childs Nerv Syst. 1997 Nov-Dec;13(11-12):608-12.
5. Grill J, Pillet P, Rakotomalala W, Andriantsimahavandy A, Esterre P, Boisier P, Guyon P. Neurocysticercosis: pediatric aspects Arch Pediatr. 1996 Apr;3(4):360-8. Review. French.
6. Riley T, White AC Jr. Management of neurocysticercosis. CNS Drugs. 2003;17(8):577-91. Review.
7. Rahalkar MD, Shetty DD, Kelkar AB, Kelkar AA, Kinare AS, Ambardekar ST The many faces of cysticercosis. Clin Radiol. 2000 Sep;55(9):668-74.
Acknowledgement
The author acknowledges the untiring work of Obidike Nwakudu, MD in the preparation of this article, while he was a resident in Family Medicine at Mount Sinai hospital, Chicago, IIinois.