


History
The patient is an elderly woman seen in a clinic because of persistent mid-back pain. She did not return to her primary care physician after this study; little of her history is available. These are the only imaging studies available in her records.
Radiologic findings
The radiographs are frontal and lateral views of her thoracic spine. They show:
1. Diffuse osteopenia.
2. Levoscoliosis of the mid and lower thoracic spine.
3. Compression fractures of the eighth and ninth thoracic vertebrae.
4. Numerous round well-defined soft-tissue masses attached to the chest wall.
The key to the radiologic diagnosis of this case lies in the non-calcified cutaneous masses of varying sizes. It is easy to assume the masses are within the patient’s thorax but a closer look shows that most are well-defined, have lucent halos around them and, so, are external to the chest wall. They are pathognomonic of von Recklinghausen’s disease (neurofibromatosis type1). The patient is osteoporotic and the vertebral compression fractures may be due to this.
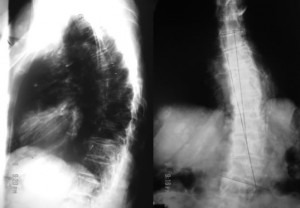
Vertebral compression fractures are common in the elderly, especially in the setting of osteoporosis. They may also occur in conditions that weaken the vertebrae such as metastatic disease, infection, Scheuermann’s disease (multiple, contiguous intraosseous herniation of nucleus pulposus at the center of weakened endplates (multiple Schmorl’snodes)), sickle-cell anemia and histiocytosis X. Again, the cutaneous masses make the diagnosis easy. Scoliosis and kyphoscoliosis of the spine may be congenital or acquired. When congenital they are due to failure of segmentation or formation of vertebral bodies; they may be mixed, showing features of both. There are many acquired causes of scoliosis and kyphoscoliosis (see below) and painful conditions and von Recklinghausen disease are two such entities. The upper and lower vertebrae in the patient show subtle signs of formation failure suggesting that in her the scoliosis is congenital (a component of the von Recklinghausen conundrum). Clearly, pain from the compression fractures may contribute to the abnormal spinal curvature. Thus, in the patient, congenital and acquired scoliosis may be at play.
Radiologic differential diagnosis
For multiple cutaneous masses:
The cutaneous masses in this patient are pathognomonic for neurofibromatosis
For scoliosis:
1. Mesodermal and neuroectodermal diseases including neurofibromatosis, Marfan’s syndrome, homocystinuria.
2. Neuromuscular diseases such as myelomeningocele, spinal muscular atrophy, Friedrich’s ataxia, poliomyelitis, cerebral palsy, and muscular dystrophy.
3. Painful scoliosis as in osteoid osteoma, osteoblastoma, intraspinal tumor, and infection.
4. Congenital scoliosis due to failure of formation, failure of segmentation, or mixed defects.
For multiple collapsed vertebrae:
1. Osteoporosis
2. Neoplastic disease
3. Scheuermann disease
4. Infection
5. Histiocytosis X
6. Sickle-cell anemia
Discussion
Neurofibromatosis type 1 (NF1) is the most common of the phakomatoses, a heterogeneous group of histiogenetic disorders characterized by the presence of central nervous system and, for the most part, cutaneous manifestations. Many also have prominent visceral and connective tissue abnormalities.
| Neurofibromatosis Neurofibromatosis type 1 Neurofibromatosis type 2 Segmental neurofibromatosis. Von Hippel-Lindau syndrome Sturge-Webber syndrome Tuberous sclerosis Others Ataxia-Telangiectasia Rendu-Osler-Weber Klippel-Trenaunay-Weber Neurocutaneous melanosis Wyburn-Masosn Basal Cell Nevus Cowden Disease. |
NF-1 occurs in one of 2000 to 3000 births. It is inherited by autosomal dominant transmission but has a high spontaneous mutation rate (50%) so that it may occur in offsprings of unaffected parents. The penetrance of the disease is high but its expressivity variable. The problem in the disease lies in the long arm of chromosome 17; there is neuroectodermal and mesodermal tissue dysplasia with potential for diffuse systemic involvement. Males and females are affected equally.
The clinical diagnosis of the disease hinges upon the demonstration of the characteristic cutaneous manifestations or two of the set of criteria shown below. The case in question has numerous cutaneous masses that are consistent with disease although we do not have a full account of the patient’s history.
The NIH Consensus Development Panel developed a set of diagnostic criteria for NF-1 that includes the following:
1. 6 or more 5mm or larger café-au-lait spots.
2. 1 plexiform neurofibroma or two or more neurofibromas of any type.
3. 2 or more pigmented iris hamartomas (so-called Lisch nodules).
4. Axillary or inguinal region freckling.
5. Optic nerve glioma
6. First-degree relative with NF-1
7. Presence of a characteristic bone lesion (e.g. dysplasia of the greater wing of the sphenoid, pseudoarthrosis).
The disease may affect many organs including the brain, spinal cord, peripheral nerves, bones, the dura, the eye and the blood vessels (see below).
| Non-CNS lesions Visceral and endocrine tumors: MEA IIb Musculoskeletal lesions: Ribbon ribs Pseudoarthroses Tibial bowing Focal overgrowth of digit, ray or limb CNS lesions Optic glioma Nonoptic glioma Basal ganglionic and white matter hamartomas Spinal cord, roots/ peripheral nerves: Hamartomas Cord astrocytoma Neurofibromas Osseous and dural lesions: Hypoplasia of the greater wing of the sphenoid bone Sutural defects Kyphoscoliosis Meningoceles Ocular/orbital manifestations: Optic nerve glioma Lisch nodules Buphthalmos Retinal phakomas |
NF-1 is different from NF-2 although it, too, is inherited by autosomal dominant transmission. NF-2 is less common than NF-1 (1:50,000), is due to defects in chromosome 22 and in it, cutaneous manifestations are rare. The dominant mode of presentation of NF-2 is by central nervous system lesions (approximately 100%). These include schwannomas of cranial and peripheral nerves, meningiomas in the brain and spine (often multiple), nonneoplastic intracranial calcifications (especially choroids plexus), spinal cord ependymomas and secondary changes of the spine.
References
1. Smirniotopoulos JG, Murphy FM: The phakomatoses, AJNR 13: 725-746, 1992.
2. Huson SM, Harper PS, Compston DA: von Recklinghausen neurofibromatosis: a clinical and population study in southeast Wales, Brain 111:1355-1381, 1988.
3. National Institute of Health Consensus Development Conference: Neurofibromatosis Conference Statement, Arch Neurol 45: 579-588, 1988
4. Diagnostic Neuroradiology, Anne G. Osborn